南京华威医药科技集团有限公司,江苏南京 210046
摘 要: 建立伊曲茶碱原料有关物质的检测方法,为其质量控制和研究提供参考依据。采用高效液相色谱法,色谱柱为ACE Super C18(250 mm×4.6 mm,5 µm),流动相A为10 mmol/l磷酸二氢钾溶液(用磷酸pH至3.0),流动相B为乙腈,进行梯度洗脱;流速为1.0 ml/min,检测波长为230 nm,柱温为40℃,进样量为10 μl。该方法的专属性好、准确度高,可采用加校正因子的主成分自身对照法对本品中的有关物质进行测定。
关键词:伊曲茶碱;有关物质;高效液相色谱法;校正因子
中图分类号:R917 文献标志码:A 文章编号:
Determination of Related Substance in Istradefylline by HPLC and Principal Component Self-control with Correction Factor
SUN Jing-long,NI Dong-sheng, BI Sen, ZHANG Yi-fan, SHAN Bing-bing, ZHAO Yi-xiang, YOU Feng
(Huawe Pharmaceutical Technology Group Co, Ltd, Nanjing 210046, China)
Abstract: To establish the detection method of itkotheophylline raw material and provide reference for its stability study and quality control. An HPLC method, A ACE Excel C18(250 mm×4.6 mm,5 µm) column was used in this study.The mobile phase A was 10 mmol/L potassium phosphate monobasic solution(adjusted to pH 3.0 with phosphoric acid) and Super was 40, the detection wavelength was 230 nm and the injection volume was 10 μl. The method has good specificity and high accuracy. It can be used to determine the related substances in this product by principal component self-control method with correction factor.
Key words: Istradefylline;Related substances;HPLC;Correction factor
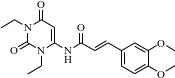
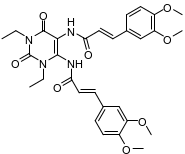
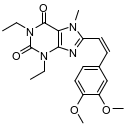
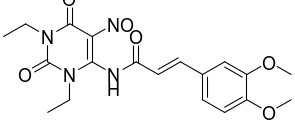
伊曲茶碱(Istradefylline)是一种新型抗PD药物,由日本Kyowa Hakko Kogyo株式会社研发,2013年5月首次在日本上市,2019年8月获美国FDA批准上市,常与多巴胺类药物联用且效果优于多巴胺类药物单独使用,具有广阔的的应用市场。其主要工艺路线是以1,3-二乙基-5,6-二氨基尿嘧啶和3,4-二甲氧基肉桂酸为原料,经酰胺化、环合、甲基化,再经重结晶纯化得到[1-2],伊曲茶碱可能存在的杂质主要有10种,合成路线及杂质结构式见图1和表1。
国内外现行质量标准中暂未收录伊曲茶碱及其制剂,目前关于伊曲茶碱原料及制剂检测的液相方法还较少,周琴芹[3]对伊曲茶碱原料药中的7个工艺杂质及降解杂质进行HPLC定量分析;吴标[4]建立了一种HPLC分析方法同时测定原料药和制剂中的8个杂质,两者均采用加校正因子的主成分自身对照法,流动相为磷酸缓冲盐和乙腈,除检测波长、pH值和洗脱梯度相差较大外,其余条件基本一致。笔者按照上述两个方法进行方法对比发现,文献[3]梯度方法可使所有杂质分开,但杂质D和J分离度小于1.0;文献[4]等度方法不能将所有杂质分开,且杂质B、C、E、H均集中在前5min出峰,现有检测方法对合成路线中潜在杂质的涵盖还较少,也还没有针对破坏实验和样品中检出的杂质进行质控限度的制定[5-6]。
综上,本文对上述两个方法进行优化,采用高效液相色谱法(HPLC法)加校正因子的主成分自身对照法对伊曲茶碱原料药中有关物质进行测定和方法学验证,以期为伊曲茶碱质量控制提供参考依据。
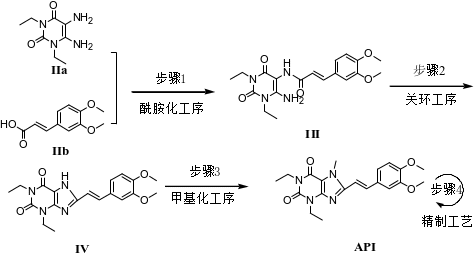 图1 伊曲茶碱合成路线
图1 伊曲茶碱合成路线
表1 各杂质结构
化合物 | 化学名称 | 结构式 | 杂质来源 |
A | (E)-1,3-二乙基-5-氢-6-(3,4-二甲氧基苯基丙烯酰基) 氨基尿嘧啶 | | 副产物 |
B | 8-[(E)-2-(3,4-二甲氧基苯基)乙烯基]-1,3-二乙基-7-氢-嘌呤-2,6-二酮 | | 中间体残留 |
C | 1,3-二乙基-5,6-二[(E)3,4-二甲氧基苯基丙烯酰基)]氨基尿嘧啶 | | 副产物 |
D | 8-[(Z)-2-(3,4-二甲氧基苯基)乙烯基]-1,3-二乙基-7-甲基嘌呤-2,6-二酮 | | 副产物、降解产物 |
E | (E)-1,3-二乙基-6-(N,N-二甲基)氨基-5-(3,4-二甲氧基苯基丙烯酰基)氨基尿嘧啶 | | 副产物 |
F | (E)-1,3-二乙基-5-亚硝基-6-(3,4-二甲氧基苯基丙烯酰基) 氨基尿嘧啶 | | 副产物 |
G | 8,8'-(2,4-双(3,4-二甲氧基苯基)环丁烷-1,3-二基)双(1,3-二乙基-7-甲基-1H-嘌呤-2,6(3H,7H)-二酮) | | 副产物、降解产物 |
H | (E)-N-(1,3-二乙基-6-(甲氨基)-2,4-二氧代-1,2,3,4-四氢嘧啶-5-基)-3-(3,4-二甲氧基苯基)丙烯酰胺 | | 副产物 |
I | 3-(3,4-二甲氧基苯基)丙烯酸酐 | | 副产物 |
J | 8-[(E)-2-(3-甲氧基-4-羟基苯基)乙烯基]-1,3-二乙基-7-甲基嘌呤-2,6-二酮 | | 降解产物 |
1 仪器与试药
伊曲茶碱对照品(批号:158190802R,纯度:99.3%)、A(批号:A+1R)、B(批号:B+3R)、C(批号:C+1R)、D(批号:D+1R)、E(批号:E+3R)、F (批号:F+1R)、G(批号:G+3R)、H(批号:H+1R)、I(批号:I+3R)、J(批号:J+2R);杂质纯度均大于90%、伊曲茶碱原料药(批号:158190801、158190811、158190821,纯度:均˃99.9%)均由本实验室自制;伊曲茶碱片(厂家:日本协和発酵キリン株式会社,批号:121AGL、123AIA,规格:20mg)。e2695型高效液相色谱仪(美国Waters公司);XPR2型百万分之一电子天平、ME204E型万分之一电子天平、S210K型pH计(瑞士梅特勒-托利多公司);DHG-9053A型鼓风干燥箱(上海一恒科学仪器有限公司);HH-S4型水浴锅(巩义市予华仪器有限责任公司);800TPS型光照箱(北京兰贝石恒温技术有限公司);TG16MW型台式高速离心机(湖南贺西仪器装备有限公司)。乙腈和磷酸为色谱纯;水为超纯水;其他试剂均为分析纯。
2 方法与结果
2.1 色谱条件
ACE Super C18色谱柱(250 mm×4.6 mm,5 µm);流动相A为10 mmol/l磷酸二氢钾溶液(用磷酸调pH至3.0),流动相B为乙腈;梯度洗脱:0~20 min,A 60%~38%;20~30 min,A 38%;30~32min,A 38%~60%;32~40min,A 60%,检测波长为230 nm,流速1.0 ml/min,柱温40℃,进样量10 μl。
2.2 溶液配制
对照品贮备溶液:(1)取伊曲茶碱对照品适量,精密称定,加流动相A-B(20:80)溶解并稀释制成每1 ml中约含50 μg的溶液,作为伊曲茶碱对照品贮备溶液。(2)取10个杂质对照品适量,精密称定,加流动相A-B(20:80)溶解并稀释制成每1 ml中约含各杂质5 μg的溶液,作为混合杂质对照品贮备溶液。
供试品溶液:(1)取伊曲茶碱原料适量,精密称定,加流动相A-B(20:80)溶解并稀释制成每1 ml中约含0.5 mg的溶液,作为原料药供试品溶液。(2)取伊曲茶碱片研碎物适量,精密称定,加流动相A-B(20:80)溶解并稀释制成每1 ml中约含0.5 mg伊曲茶碱的溶液,离心,取上清液作为制剂供试品溶液。
自身对照溶液:(1)精密量取原料药供试品溶液1.0 ml,置于100 ml量瓶中,加流动相A-B(20:80)稀释至刻度,摇匀,作为原料药自身对照溶液。(2)精密量取制剂供试品溶液1.0 ml,置于100 ml量瓶中,加流动相A-B(20:80)稀释至刻度,摇匀,作为制剂自身对照溶液。
系统适用性溶液:取伊曲茶碱对照品贮备溶液及混合杂质对照品贮备溶液适量,精密称定,加流动相A-B(20:80)溶解并稀释制成每1 ml中约含伊曲茶碱0.5 mg和各杂质0.5 μg的溶液,作为系统适用性溶液。
空白溶液及空白辅料溶液:(1)以流动相A-B(20:80)作为空白溶液。(2)取处方量空白辅料适量,精密称定,加流动相A-B(20:80)溶解并稀释制成每1 ml中约含3 mg辅料的溶液,离心,取上清液作为空白辅料溶液。
2.3 系统适用性试验
精密量取“2.2”项下的系统适用性溶液,按“2.1”项下色谱条件进样分析,记录色谱图。结果伊曲茶碱峰与各杂质峰间无干扰,分离度均>1.5,理论板数以主成分峰计>5000。系统适用性图谱见图2。
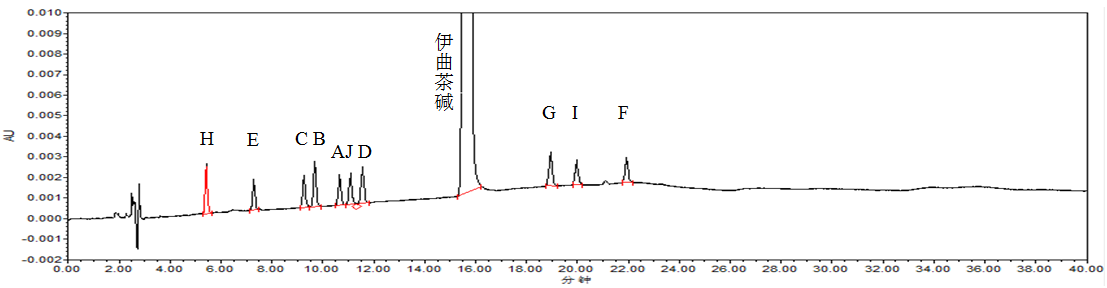
 图2 系统适用性试验色谱图
图2 系统适用性试验色谱图
2.4 强制降解试验
精密称取原料供试品(批号:158190801,下同)约25 mg,共8份,分别置于50 ml量瓶中,1份为未破坏样品,另7份按以下条件进行破坏。(1)酸破坏试验:加入1.0 mol/l盐酸溶液2 ml,室温放置10天后,加1.0 mol/l氢氧化钠溶液2 ml中和,加流动相A-B(20:80)稀释至刻度,摇匀。(2)碱破坏试验:加入1.0 mol/l氢氧化钠溶液2 ml,室温放置10天后,加1.0 mol/l盐酸溶液2 ml中和,加流动相A-B(20:80)稀释至刻度,摇匀。(3)氧化破坏试验:加入30%双氧水5.0 ml,室温放置10天,加流动相A-B(20:80)稀释至刻度,摇匀。(4)高温固体破坏试验:105℃高温破坏10天,放冷后,加流动相A-B(20:80)溶解,稀释至刻度,摇匀。(5)高温液体破坏试验:加流动相A-B(20:80)适量使溶解,105℃高温破坏10天,加流动相A-B(20:80)稀释至刻度,摇匀。(6)光照固体破坏试验:4500 Lx光强下照射9天,加流动相A-B(20:80)溶解,稀释至刻度,摇匀。(7)光照液体破坏试验:加流动相A-B(20:80)适量使溶解,4500 Lx光强下照射5分钟,加流动相A-B(20:80)稀释至刻度,摇匀。取上述8份供试品溶液,按“2.1”项下色谱条件进样分析,记录色谱图。结果,本品在酸碱、氧化和高温条件下较稳定;在光照液体和固体条件下均可检出降解产物,经结构确证为杂质D和G,降解产物峰与主峰均能达到基线分离,且各峰间分离度良好,见图谱3。
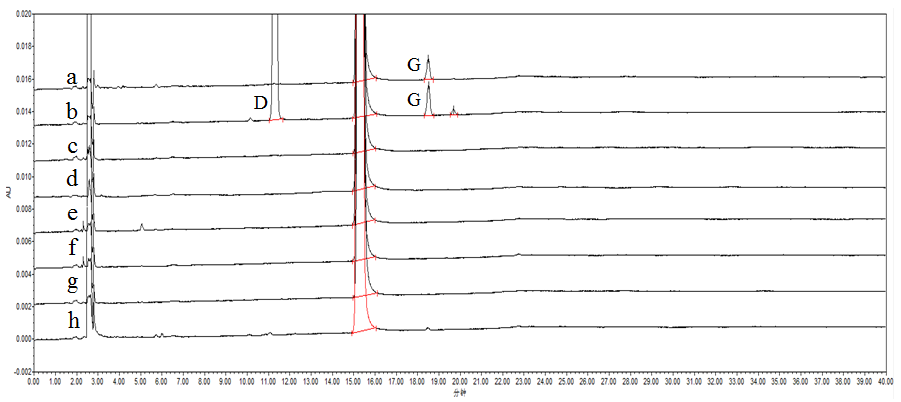 图3 强制降解溶液色谱图
图3 强制降解溶液色谱图
a-光照固体破坏;b-光照液体破坏;c-未破坏;d-高温固体破坏;e-高温液体破坏;f-酸破坏;g-酸破坏;h-氧化破坏
2.5 线性关系及校正因子试验
分别精密量取“2.2”项下对照品贮备溶液适量,加流动相A-B(20:80)稀释制成每1 ml中含伊曲茶碱为0.125、0.25、0.50、0.75、2.50、5.00、6.00 μg的系列溶液,加流动相A-B(20:80)稀释制成每1 ml中含杂质A~J均为0.125、0.25、0.50、0.75、1.00、1.25 μg的系列溶液。分别按“2.1”项下色谱条件进样分析,记录色谱图。以伊曲茶碱或各杂质质量浓度为横坐标(X),以峰面积为纵坐标(Y),进行线性回归,并以方程斜率(K)计算各杂质的校正因子(校正因子=K伊曲茶碱/K杂质),杂质D和G计算时乘以校正因子0.8,其余杂质计算时校正因子均不计[7],结果见表2。
表2 线性关系及校正因子
化合物 | 范围(µg/ml) | 线性方程 | r | 校正因子 |
A | 0.12~1.17 | Y= 2.7861×104X- 2.19×102 | 1.0000 | 0.95 |
B | 0.13~1.27 | Y= 2.7829×104X- 7.70×101 | 0.9999 | 0.96 |
C | 0.12~1.19 | Y= 2.6683×104X+ 3.95×102 | 0.9999 | 1.00 |
D | 0.11~1.14 | Y= 3.2006×104X- 1.99×102 | 0.9999 | 0.83 |
E | 0.12~1.16 | Y= 2.3768×104X+ 2.54×102 | 1.0000 | 1.12 |
F | 0.11~1.15 | Y= 2.3456×104X- 1.09×102 | 0.9999 | 1.13 |
G | 0.13~1.32 | Y= 2.7019×104X+ 8.91×101 | 0.9999 | 0.98 |
H | 0.11~1.12 | Y= 3.1572×104X- 1.03×102 | 1.0000 | 0.84 |
I | 0.13~1.26 | Y= 2.6875×104X- 8.68×101 | 0.9999 | 0.99 |
J | 0.12~1.21 | Y= 2.6716×104X+ 2.19×102 | 0.9999 | 0.99 |
伊曲茶碱 | 0.13~6.0 | Y= 2.6577×104X- 2.42×102 | 1.0000 | / |
2.6 精密度试验
取“2.5”项下质量浓度为0.75 μg/ml的杂质溶液和0.75 μg/ml的伊曲茶碱溶液,分别按“2.1”项下色谱条件连续进样6次,记录色谱图。结果,伊曲茶碱及各杂质峰面积的RSD均小于1.8%(n=6),表明该方法精密度良好。
2.7 检测限与定量限考察
取“2.2”项下对照品贮备溶液,逐级稀释,按“2.1”项下色谱条件进样分析,记录色谱图。以信噪比为3:1计算检测限,以信噪比为10:1计算检测限,结果,伊曲茶碱及杂质A~J的检测限分别为48、47、51、48、46、46、46、53、45、50、48 ng,定量限分别为120、117、127、119、114、116、115、132、112、126、121 ng。
2.8 稳定性试验
取“2.2”项下原料供试品溶液、原料自身对照溶液、系统适用性溶液适量,在室温避光下分别放置0、4、8、16、24 h后,按“2.1”项下色谱条件进样分析,记录色谱图。结果,原料供试品溶液中伊曲茶碱峰面积RSD为0.20%(n=5);原料自身对照溶液中伊曲茶碱峰面积RSD为0.15%(n=5);系统适用性溶液中伊曲茶碱峰面积RSD为0.52%(n=5)、各杂质峰面积的RSD均小于1.5%(n=5),表明原料供试品溶液、原料自身对照溶液、系统适用性溶液在室温避光下放置24 h内稳定性良好。
2.9 重复性试验
取原料供试品,按“2.2”项下方法制备重复性供试品,平行6份,按“2.1”项下色谱条件进样分析,记录色谱图。结果,杂质A~J均未检出;表明该方法的重复性良好。
2.10 加样回收率试验
精密称取原料供试品约25 mg,共9份,各置于50 ml量瓶中,分别按供试品溶液浓度的0.05%、0.15%、0.225%的量加入“2.2”项下混合杂质对照品贮备溶液,加流动相A-B(20:80)稀释至刻度,摇匀,制得低、中、高3种浓度的回收率试验溶液,各3份。精密量取各回收率试验溶液1.0 ml,置于100 ml量瓶中,加流动相A-B(20:80)稀释至刻度,摇匀,作为自身对照溶液。按“2.1”项下色谱条件进样分析,记录色谱图。按“2.5”项下校正因子,采用加校正因子的自身对照法计算各杂质加样回收率。结果,杂质A~J平均加样回收率分别98.2%、99.8%、98.4%、96.9%、99.5%、98.3%、99.1%、97.8%、98.4%、99.4%,RSD分别为0.83%、1.8%、1.8%、1.8%、1.7%、1.1%、1.5%、0.84%、1.3%、1.2%(n=9),表明该方法的准确度良好。
2.11 耐用性试验
精密量取“2.2”项下的系统适用性溶液,以各色谱峰的分离度为指标,分别考察同型号不同批号色谱柱、检测波长(228和232 nm)、柱温(38和42℃)、流速(0.9和1.1 ml/min)、流动相pH值(2.8和3.2)等条件变化对检测结果的影响。结果,当以上条件发生变化时,各色谱峰均可以满足系统适用性试验的要求,该方法耐用性良好。
2.12 样品测定
取原料药(批号:158190801、158190811、158190821)3批和原研制剂(121AGL、123AIA)2批,分别按“2.2”项下方法制备供试品溶液、自身对照溶液、空白溶液及空白辅料溶液,按“2.1”项下色谱条件进样分析,记录色谱图。按“2.5”项下校正因子,采用加校正因子的自身对照法计算各杂质含量[10]。结果,原料药3批供试品中仅有158190811批次检出杂质B,含量为0.03%,其余杂质均未检出;原研制剂2批供试品中仅有123AIA批次检出杂质G,含量为0.03%,其余杂质均未检出。
3 讨论
原料药仅检出杂质B,原料破坏条件(光照固体和液体)下主要降解出杂质D和G,原研制剂检出杂质G。伊曲茶碱为8-苯乙烯基黄嘌呤衍生物,在稀溶液光照状态下会发生顺反异构化生成杂质D、在固体光照状态下发生环加成生成杂质G,因此在固液状态下均应考虑其光敏性[8],综上所述,伊曲茶碱原料药在存储和实验样品配制过程中应避光。伊曲茶碱片原研说明书规定药品规格为20 mg/片,每日2次,按最大服用量计算每日最大剂量为40 mg。根据指导原则[9-10]建议,结合原料和制剂的检测结果拟定限度:杂质D、B、G均不得超过0.15%,其他单个杂质不得超0.10%,总杂不得超过0.5%。
本研究采用HPLC法测定伊曲茶碱中有关物质,所建立方法专属性强,精密度、准确度均良好;为该品种的质量控制、贮藏条件等提供了参考。
参考文献:
[1] 龚登凰, 王洁, 胡瑞. 伊曲茶碱的合成工艺改进[J]. 精细化工中间体, 2015, 45(02): 26-29.
[2] 齐军阳. 伊曲茶碱的合成研究[D]. 湖北: 武汉工程大学, 2015: 18-23.
[3] 周琴芹. 伊曲茶碱原料药的有关物质研究[D]. 江苏: 南京工业大学, 2015: 04-33.
[4] 吴标, 崔红晓, 施伶俐, 等. 一种伊曲茶碱有关物质的高效液相色谱分析方法[P]. 2016: 01-12.
[5] 王维剑. 药品杂质控制与评价关键技术研究[D]. 山东: 山东大学, 2016: 10-20.
[6] 胡昌勤. 化学药品杂质谱控制的现状与展望[J]. 中国新药杂志, 2015, 24(15): 1721-1731.
[7] 吴琼, 宋丽丽. 丙氨酰谷氨酰胺9个有关物质的UPLC法测定[J]. 中国医药工业杂志, 2019, 50(9): 1042-1046.
[8] Hockemeyer J , Burbiel J C , Müller, Christa E. Multigram-Scale Syntheses, Stability, and Photoreactions of A 2A Adenosine Receptor Antagonists with 8-Styrylxanthine Structure:? Potential Drugs for Parkinson\"s Disease[J]. Journal of Organic Chemistry, 2004, 69(10): 3308-3318.
[9] 国家药品审评中心. 化学药物杂质研究技术指导原则[EB/OL]. (2007-08-23)
[10] ICH Harmonised Tripartite Guideline: Impurities In New Drug Substances, Q3A(R2)[s]. 2006.
1作者简介:孙井龙,男,硕士研究生,主要从事药品分析与质量研究。E-mail:sjlsjlong@126.com。

客服QQ:30444492琼网文【2021】1550-113号
增值电信业务经营许可证:琼B2-20210322
出版物经营许可证:新出发龙华出字第(2021)009号
广播电视节目制作经营许可证:(琼)字第00779号
版权所有 ©2002-2024 期刊网 琼ICP备2021005105号