陕西理工大学化学与环境科学学院 汉中 723000
摘要 采用量子力学中CBS-QB3的组合方法对2-甲基呋喃与OH自由基的反应机理进行了理论计算研究。研究结果表明,标题反应存在三类反应,分别是加成反应,SN2取代反应和抽氢反应,共有10条通道。在加成反应中,主要发生在C1、C2、C3和C4上,吉布斯自由能变化值均小于零,分别为-114.68,-36.54,-41.73,-116.81 kJmol-1,表明该类反应从热力学角度分析均可进行;四条加成反应活化能分别为0.00,24.40,15.62,0.00 kJmol-1,产物稳定性为P4(C4)>P1(C1)>P3(C3)>P2(C2),表明四条加成反应无论从动力学还是热力学均可容易进行,其中OH极易加成在C1,C4位置上,是加成反应的优势通道。在SN2取代反应中,吉布斯自由能变化值为93.34 kJmol-1 ,反应活化能为201.58 kJmol-1,可见SN2取代反应从热力学和动力学角度分析,均不易进行。抽氢反应有5条反应通道,分别抽取2-甲基呋喃环上和甲基上氢,反应吉布斯自由能变化值分别为16.52,17.48,18.28,-122.89,-122.26 kJmol-1,表明OH自由基抽取H2、H3、H4反应通道从热力学角度不易进行。OH自由基抽取H6和H7反应活化能分别17.74,17.75 kJmol-1,且抽氢产物P9和P10产物也极为稳定。因此OH自由基抽取甲基上H原子形成的类似卞基结构的产物P9,P10的反应为抽氢反应的优势通道。
关键词 2-甲基呋喃 OH自由基 抽氢反应 SN2取代反应 加成反应
中图分类号: O647.5 文献标识码:A 文章编号:
Theoretical Study on the 2-methyl Furan and Hydroxyl Radical*
Chenyang Che Zixuan Yang Mingfeng Ying RuiRui Li Mengdan Lv
School of Chemical & Environment Science, Shaanxi University of Technology, Han zhong 72300, China
Abstract: The mechanism of 2-methylfuran with hydroxyl radical is studied by using the CBS-QB3 method. Theoretical research results show that there are three title reaction ways, such as addition, SN2 substitution, and hydrogen abstraction, respectively. There are altogether ten reaction channels. As for the addition, the reaction mainly occurs in C1, C2, C3 and C4, the change values of the Gibbs free energies are less than zero, which are -114.68, -36.54, -41.73, -116.81 kJmol-1, respectively, indicating that the addition reaction can be carried out in thermodynamics; The activation energies of the addition paths are 0.00, 24.40, 15.62, 0.00kJmol-1and the stability of the product is P4(C4)>P1(C1)>P3(C3)>P2(C2), which indicates that the four addition reactions could be easily carried out in both kinetics and thermodynamics. The OH radicals are easy to be added to C1 and C4 positions and are the dominant channel of addition reaction. As for SN
2 substitution, the change value of the Gibbs free energy is 93.34 kJmol-1 and the activation energy is 207.88 kJmol-1. Therefor, the SN2 substitution is very difficult to react in kinetics and thermodynamics. As for the hydrogen abstraction reaction, there are mainly five reaction channels. The change values of the Gibbs free energies are 16.52, 17.48, 18.28, -122.89, -122.26kJmol-1, respectively. These indicate that the abstraction of H2, H3 and H4 atoms are difficult to extract in thermodynamics. The activation energies of the abstraction of H6, H7 atoms are 17.74, 17.75 kJmol-1, respectively. The P9 and P10 are most stable in all hydrogen products. Therefore, the abstraction of H6 formed P9 which is similar to the bienyl structure is the dominant channel.
Keywords: 2-methylfuran; hydroxyl radical; addition reaction; SN2 substitution reaction; hydrogen abstraction
1 引言
化石燃料和生物物质的燃烧造成的环境问题日益严重,其中含氧、氮,硫的杂环有机化合物对人类健康已经产生了较大的威胁[1,2]。呋喃是一种具有高挥发性和亲脂性的芳香族环醚,主要是通过生物质燃烧释放到大气中,对空气质量及人类健康存在潜在危害。关于呋喃及其衍生物的相关研究,一直是人们关注的焦点,其中2-甲基呋喃和2,5-二甲基呋喃,由于特定的物理性质和化学性质,在减少生物柴油燃烧产生的烟尘排放及替代相关燃料方面应用较为广泛[3]。OH自由基具有较强的氧化能力,主要产生于臭氧光解、气态亚硝酸光解等反应以及HO2自由基与NO或O3的反应过程中。在研究水污染,大气光化学污染及生物医学基因表达及细胞生化代谢过程中扮演着不可替代的角色[4-6]。
据报道,E. Gómez Alvarez、E. Borrása等人通过研究2-甲基呋喃、3-甲基呋喃与OH自由基的反应指出1,4-二羰基化合物是呋喃及其衍生物与OH自由基反应的主要产物,并得出结论OH自由基与呋喃及其衍生物之间主要存在着三种类型的反应,分别是抽氢反应、SN2反应和加成反应[7]。Whelan等人在不同压力和温度下对OH自由基与呋喃及其呋喃衍生物2-甲基呋喃,2,5-二甲基呋喃的动力学反应进行了相关研究[8],但目前关于OH自由基与2-甲基呋喃,2,5-二甲基呋喃的理论计算研究却鲜有报道。
鉴于呋喃及其衍生物在大气污染治理和人类生活中的重要地位,本文采用量子化学CBS-QB3组合方法[9]对2-甲基呋喃与OH自由基的反应机理进行详细深入研究,分别从热力学和动力学角度揭示其反应规律,为实验工作者在呋喃及其衍生物的降解、消除及生物医学研究等过程中提供更多具有参考价值的信息。
2 计算方法
本文选用量子化学中CBS-QB3的组合方法,分别对2-甲基呋喃与OH自由基的加成,SN2取代和抽氢反应过程中的各驻点进行了计算,获得了各个反应的反应活化吉布斯自由能∆G≠以及吉布斯自由能变化值的∆G,相对吉布斯自由能GR,绘制出了势能面剖面图。采用B3LYP/6-311G(d,p)[10]对所有驻点包括反应物复合物、过渡态及产物构型进行了全优化,并在相同水平上进行了频率计算和内禀反应坐标(IRC)[11]分析,以确认各驻点的稳定性和反应路径连接的合理性。其中CBS-QB3是一种高精度的组合方法,先优化再能量计算五步组成:采用B3LYP/6-311G(d,p)方法进行几何构型优化以及频率计算,然后采用CCSD(T)、MP4SDQ、MP2及CBS外推法进行能量校正。
3 结果与讨论
采用CBS-QB3组合方法对这三类反应分别进行了理论研究,图1示出了在B3LYP/6-311G(d,p)方法水平上优化得到的各反应物、中间体、过渡态以及产物的几何构型。图2绘出了反应在该组合方法水平上的势能剖面图,直观地反映了反应过程中能量的变化。表1列出了反应通道各驻点物种的反应活化吉布斯自由能
∆G≠,吉布斯自由能变化值∆G,相对吉布斯自由能GR,为分析各类反应提供了可靠的依据。
3.1 加成反应通道
OH自由基具有高反应活性,可与2-甲基呋喃不饱和原子C1,C2,C3,C4位置发生加成反应,用R1~R4分别进行标记,加成产物分别记为P1~P4。从表1各物种的能量信息中可知P1~P4的相对能量分别为-114.68,-36.54,-41.73,-116.81 kJmol-1,其稳定顺序P4(C4)>P1(C1)>P3(C3)>P2(C2),由此可知OH自由基在C1和C4位置的加成形成P1和P4有较高的稳定性。
OH自由基参与的加成反应,在大多数情况下,其势能面较为平缓,不易确定过渡态结构。采用TS,QST2以及QST3等多种方法对OH自由基与C1,C2,C3,C4位置的加成反应过渡态进行了寻找,均未能找到OH在C1及C4位置加成的过渡态结构。因此,我们以OH自由基的O原子与2-甲基呋喃中的C1和C4原子间的距离L为变量,在B3LYP/6-311G(d,p)水平上分别对2个加成反应进行了松散势能面扫描,结果如图3所示。显然,在R1和R4能量曲线上,均无能量最高点,表明OH自由基与C1和C4位置的加成生成P1和P4是个无势垒反应过程。加成反应分子结构图1和势能面剖面图2也证实了OH在C1和C4位置的加成为无势垒快速自发过程,是OH与2-甲基呋喃加成反应的主通道。
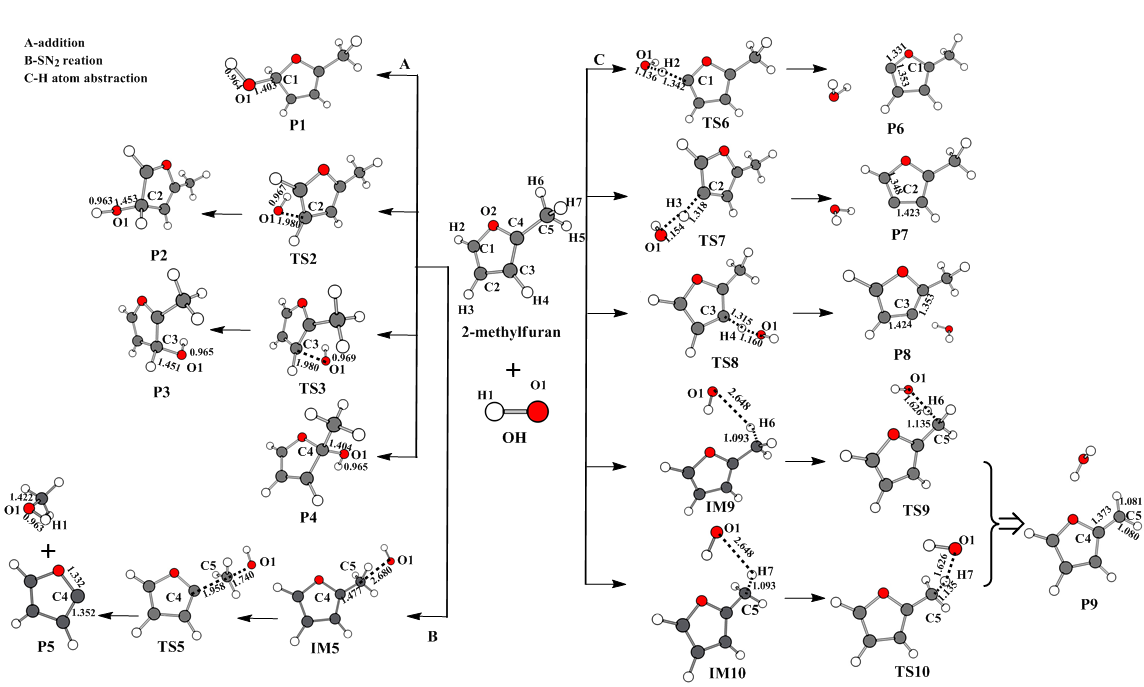
图1 标题反应在CBS-QB3组合方法水平上优化所得的各反应物、产物、中间体及过渡态的几何构型 [单位:键长(Å)]
Fig.1 Geometric configurations of reactants, products, intermediates, and transition states optimized at the level of CBS-QB3 combination method [unit: bond length (Å)]
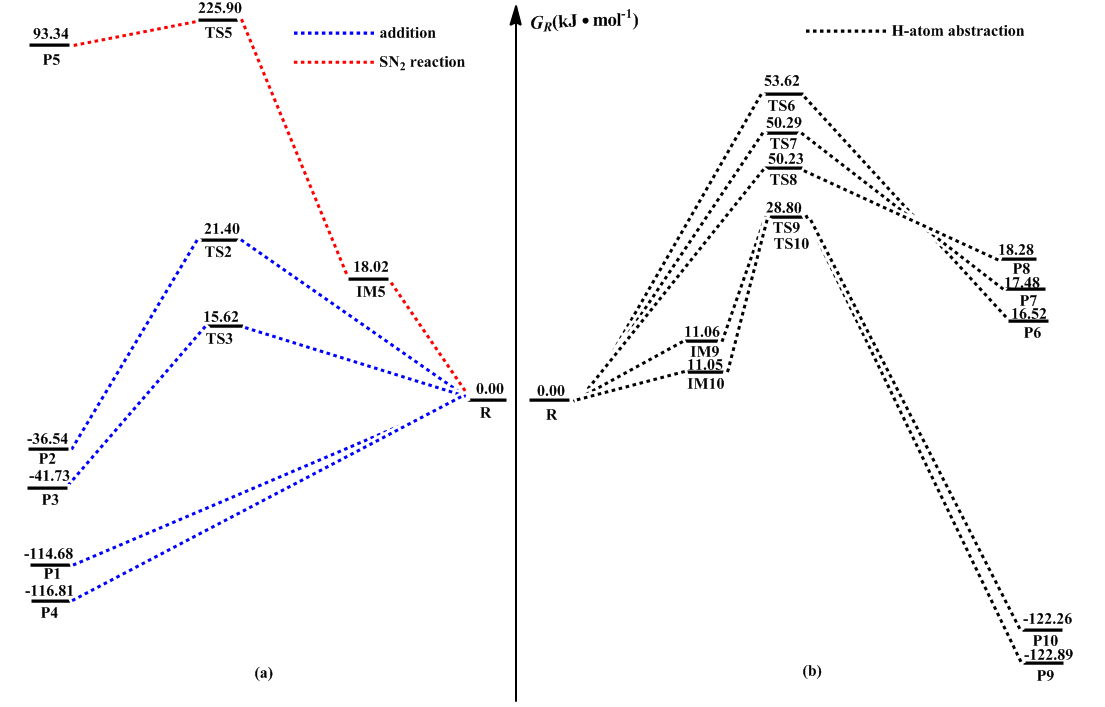
图2 标题反应在CBS-QB3组合方法水平的势能剖图
Fig.2 The potential Energy profile of the title response at the level of the CBS-QB3 combination method
图
3 OH自由基与2-甲基呋喃在C1和C4位置的加成反应势能面扫描
Fig. 3 The relax scan of potential energy surface for the OH radical add to the C1 and C4 sites of 2-methylfuran.
优化OH自由基与C2和C3位置的加成反应通道,过渡态对应的构型仅有一个虚频,分别为-333.94, -334.28 i cm-1,对虚频对应振动模式分析及IRC验证,均表明TS2和TS3确实是势能面上的一阶鞍点,是加成反应合理的过渡态。从表1可以看出,反应物R经过渡态TS2生成P2需要21.40 kJmol-1的能垒;而在反应物R经过渡态TS3生成产物P3的过程中,仅仅需要15.62 kJmol-1的能垒,显然生成P3的几率要远高于P2。
3.2 SN2取代反应通道
将OH自由基与2-甲基呋喃在C5之间发生的双分子亲核SN2背面取代反应记为R5,从表1中可知吉布斯自由能变化值为93.34 kJmol-1,反应活化能为201.58 kJmol-1 ,显然,该反应在热力学和动力学上是不容易进行的。具体反应历程为:2-甲基呋喃与OH自由基放出18.02 kJmol-1的热量形成IM5中间体,再经过207.88 kJmol-1的高能垒形成过渡态TS5,随后甲基从呋喃环上脱落,生成产物P5和自由基+CH3OH。具体反应历程见图1。
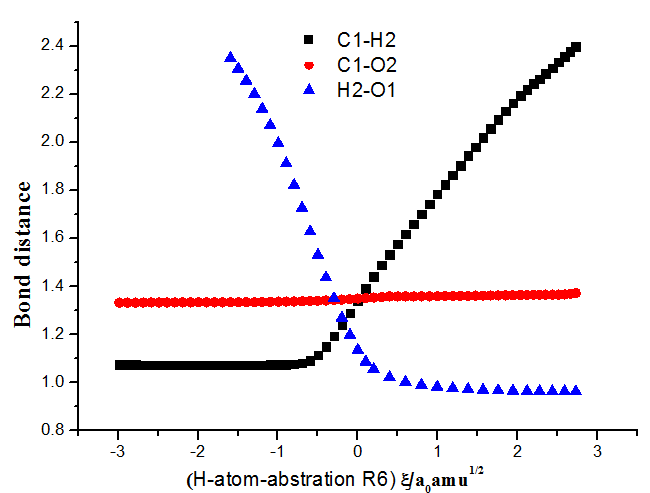
图4 抽氢反应通道R6相关键长随反应的变化
Fig.4 The change of bond length for R6 along the reaction coordinates.
3.3 抽氢反应通道
将OH自由基抽取2-甲基呋喃环上碳原子的H2,H3和H4,甲基上的H6,H7的反应分别记为R6~R10,具体反应历程见图1。反应开始时,OH自由基可分别抽取2-甲基呋喃环上的C1,C2,C3上的氢原子,在CBS-QB3组合方法上进行优化得到了反应历程中过渡态TS6,TS7,TS8和产物P6,P7,P8的几何构型,分别生成产物自由基P6+H2O,P7+H2O,P8+H2O。本文以反应通道R6为例绘制出了键长随反应进行的变化趋势,如图4所示。从表1中可知这三条反应通道对应的吉布斯自由能变化值分别是16.52,17.48,18.28 kJmol-1,可见2-甲基呋喃环上的C1,C2,C3的抽氢反应在热力学上是不容易进行反应的。
此外,OH自由基的氧原子可以抽取2-甲基呋喃甲基上的氢原子,由于甲基上的3个H近似等价,其空间结构为sp3杂化,分子在空间中容易转动,导致反应R9与R10生成类似产物。以通道R9为例进行讨论,反应物经历一个放热过程生成中间体IM9,接着需要越过17.74 kJmol-1的能垒生成TS9,最后生成产物自由基P9+H2O。从表1中可得到这两条反应通道对应的吉布斯自由能变化值分别为-122.89,-122.26 kJmol-1,反应活化吉布斯自由能垒分别为17.74,17.75kJmol-1,由此可知该反应通道在热力学和动力学上均可发生的。
通过上述分析可以看出,OH自由基抽取甲基上的H比抽取2-甲基呋喃环上C原子上的H更容易,且形成更稳定的类苄基化合物。因此,体系中生成产物P9,P10的反应是抽氢反应的优势通道。
4 结论
本文采用CBS-QB3组合方法对2-甲基呋喃与OH自由基的反应机理进行了理论研究,结果表明:
在加成反应的4条通道中,通道R1,R2,R3,R4吉布斯自由能变化值均小于零,反应活化吉布斯能均较低,尤其R1,R4为无势垒反应,且形成的加成产物也较为稳定,因此R1和R4反应为加成反应的优势通道。
SN2取代反应通道R5中吉布斯自由能变化值和反应活化吉布斯自由能垒极高,因此从热力学和动力学角度分析均不容易上进行。
在抽氢反应的5条通道中,甲基上发生的抽氢反应R9,R10的吉布斯自由能变化值和反应活化吉布斯自由能均较小,且形成更稳定的类苄基化合物,因此R9,R10为抽氢反应的优势通道。
总之,以上结果为实验工作者研究2-甲基呋喃与OH自由基反应机理提供了一定的理论参考。
参考文献
Y Zhang, F Bao,M Li. Environ. Sci. Technol., 2020, 54(23):14868-14876.
S Meng, S Boxiong, A George, F X Li, J L Li, Y Peng, P G Hong, L Cai, H G Qi. J. Environ. Sci., 2021,101(03): 49-71.
Alexandrino K. Energy Fuels., 2020, 34(6): 6598-6623.
杨新平, 王海潮, 谭照峰. 化学学报, 2019, 77(07): 41-52.
Liu Q, Liggio J, Wentzell J J B. Environ. Sci. Technol. Lett., 2020, 7(9): 646–652
张桂玲, 戴柏青. 中国生物化学与分子生物学报, 2000(01):120-123.
Tapia A , Villanueva F, Salgado M S. Atmos. Chem. Phys., 2011, 10(10): 3227-3241.
Whelan C A, Eble J, Mir Z S. J. Phys. Chem. A., 2020, 124(37): 7416-7426.
Ess D H, Houk K N. J. Phys. Chem. A., 2005, 109(42):9542-9553.
Sirjean B, Fournet, Rene. J. Phys. Chem. A., 2012, 116(25):6675-6684.
Cabaas B, Villanueva F, P. Martin. Atmos. Environ., 2005, 39(10):1935-1944.
收稿日期: ; 修回日期:
基金项目:陕西理工大学“大学生创新创业训练计划项目”项目(S202110720045)资助
联系人简介:吕梦丹(1994-),女,硕士研究生,主要研究方向:理论计算化学。Email:lvmengdan0208@126.com

客服QQ:30444492琼网文【2021】1550-113号
增值电信业务经营许可证:琼B2-20210322
出版物经营许可证:新出发龙华出字第(2021)009号
广播电视节目制作经营许可证:(琼)字第00779号
版权所有 ©2002-2024 期刊网 琼ICP备2021005105号



