(西南大学 化学化工学院, 重庆 400715)
摘要:本文报道了一种新型负载型双催化剂的模型,该模型可以解决昂贵均相催化剂浪费的问题。
关键词:高分子多孔材料;双催化
自从20世纪60年代有机化学家报道了最初几例金属配合物(金属有机化合物)催化的不对称反应以来[1],不对称金属催化得到了迅猛发展,成为有机合成方法学的重要组成部分[2]。不对称有机催化最早出现于20世纪初期[3,4],虽然早于金属催化的不对称反应,但直到本世纪才得到快速发展,现已经成为金属不对称催化和酶催化之外的第三种催化对映选择性合成手性分子的重要方法[5]。根据活化底物模式的不同,有机小分子催化主要分为烯胺催化、亚胺离子催化、布朗斯特酸碱催化、亲核催化和相转移催化等。从活化模式可以看出,有机小分子催化主要适用于官能团化的有机分子,有机催化使更广泛的官能团经历不同范围的对映选择性转化,对于只含有非活泼化学键的分子,有机小分子催化剂通常难以活化。金属催化剂往往能够实现这类底物的活化。因此,需要惰性键的活化或功能化的有机转化需要使用金属催化,这使得化学键的范围比有机催化所允许的更广泛得多。在过去的二十年里,一种主要是将过渡金属催化和有机催化结合起来的新方法已经成为一个新兴的研究领域。
2.1实验仪器和试剂
仪器:扫描电子显微镜,投射电子显微镜,比表面仪,高效液相色谱仪,Av-600型傅立叶变换器超导核磁共振仪(Bruker公司瑞士),SHB型真空水泵,LOOYZX98-1旋转蒸发仪,85-2型恒温磁力搅拌器等。
试剂与原料:1,2-二氯乙烷(DCE);氯甲基甲醚; 二(三苯基膦)二氯化钯 [(C6H5)3P]2PdCl2(98%,毕得医药);手性联萘磷酸TRIP(99%,毕得医药)。
2.2催化剂合成步骤
高分子载体的制备:我们以苯乙烯St为单体进行聚合反应的得到高分子聚苯乙烯纳米载体,在制备过程中我们我加入了足量的造孔剂,以确保我们的载体有足够的介孔形成,我们以无水乙醇溶液作为相分散剂去分散苯乙烯单体,之后我们加入90 mg 过硫酸钾KPS、蒸馏水( 8 mL ),以过硫酸钾为聚合反应引发剂去启动乳液聚合,随后我们又继续滴加无水乙醇, AIBN对其高分子聚苯乙烯进行交联聚合,以确保后面对其形貌的稳定,保持温度在80 ℃ 以上进行反应60 min。我们将反应置于80 ℃以上反应36 h。最后离心固体用四氢呋喃对其进行刻蚀确保线性的聚苯乙烯被消除得到我们空心的高分子纳米模型。
催化剂的锚定:我们将上部所制备的高分子纳米模型干燥后取部分材料作为催化剂锚定的载体,采用 DCE溶液作为溶剂对高分子纳米模型进行分散搅拌,室温搅拌均匀后加入少量MOMCl和FeCl3作为氯甲基试剂,在我们聚苯乙烯的苯环上进行氯甲基化方便后续催化剂的负载,反应一段时间后我们继续加入一个含有机小分子催化剂手性联萘磷酸和过渡金属催化剂[(C6H5)3P]2PdCl2的溶液,随后我们将反应置于55 ℃油浴锅中,反应50 h,反应结束,离心后我们将固体置入真空干燥箱中干燥得到我们的负载型双催化剂棕色固体。
2.3催化反应探究
我们以2-苯基丙醛与3-苯基烯丙基醇,为模板反应,苯为溶剂,加入我们所制备负载型高分子空心模型双催化剂,进行反应,成功得到了目标产物,并且有88 % 的产率和80 % 的ee值。

88% yield, 80% ee
表1 不同催化剂对模板反应的探究
a除非特别声明,反应条件为:在反应管中依次加入[(C6H5)3P]2PdCl2(5 mol%),TRIP (10 mol%),aldehyde (0.2 mmol), (allyl alcohol 0.2 mmol), benzhydrylamine (40 mol%),最后加入Dry benzene(1 mL),密封反应管,室温搅拌5分钟,随后于50oC油浴锅中反应12h;a经柱层析纯化后的产率;b经HPLC手性柱子后的ee值
2.4目标产物核磁数据
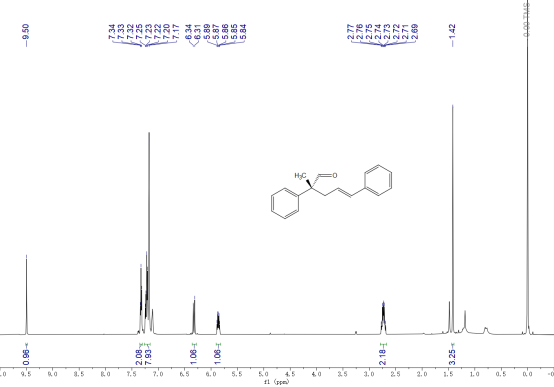
 1H NMR (600 MHz, CDCl3) δ 9.50 (s, 1H), 7.33 (t, J = 7.6 Hz, 2H), 7.26 – 7.15 (m, 8H), 6.32 (d, J = 16.9 Hz, 1H), 5.86 (dt, J = 16.0, 7.5 Hz, 1H), 2.73 (qd, J = 14.2, 7.5 Hz, 2H), 1.42 (s, 3H).
1H NMR (600 MHz, CDCl3) δ 9.50 (s, 1H), 7.33 (t, J = 7.6 Hz, 2H), 7.26 – 7.15 (m, 8H), 6.32 (d, J = 16.9 Hz, 1H), 5.86 (dt, J = 16.0, 7.5 Hz, 1H), 2.73 (qd, J = 14.2, 7.5 Hz, 2H), 1.42 (s, 3H).
参考文献
[1](a) NozakiH,MoriutiS,TakayaH,NoyoriR.TetrahedronLett.,1966,7:5239.;(b)NozakiH,TakayaH,MoriutiS,NoyoriR.Tetrahedron,1968,24:3655.;(c)KnowlesWS,SabackyMJ, Chem.Commun.,1968,22:1445.;(d)HornerL,SiegelH,BütheH.Angew.Chem.Int.Ed.,1968,7:942.
[2](a)JacaobsonEN, PfalatzA, YamamotoH. ComPrehensive AsymmetricCatalysis. SPringer-Verlag: Heidelberg, 1999.; (b) Zhou Q L. Privileged Ligands and Catalysis, Willey-VCH: Weinheim, 2011.; (c) Halpem J, Trost B M. Pure. Natl. Aead. Sci., 2004, 101:5347.
[3]BredigG,FiskeWS.Biochem.Z.,1912,46:7.
[4]MacMillanDWC.Nature,2008,455:304.
[5](a)DalkoPI,MoisanL.Angew.Chem.Int.Ed.,2001,40:3726.;(b)DalkoPI.EnantioselectiveOrganocatalysis.Wiley-VCH:Weinheim,2007.;(c)ListB.Chem.Rev.,2007,107:5413.
附录产物谱图

客服QQ:30444492琼网文【2021】1550-113号
增值电信业务经营许可证:琼B2-20210322
出版物经营许可证:新出发龙华出字第(2021)009号
广播电视节目制作经营许可证:(琼)字第00779号
版权所有 ©2002-2024 期刊网(www.qikanchina.com) 琼ICP备2021005105号



