成都蓝蜻蜓生物技术有限公司 成都市 610000
摘要
仑伐替尼是一个具有多靶点的小分子酪氨酸酶抑制剂,尤其对VEGFR有更强的抑制活性,其市场反应较好,但存在一定的缺陷。本文对仑伐替尼进行一定的机构优化,开发了两种仑伐替尼的衍生药物并对其进行体外活性研究,同时详细描述了该类衍生药物的合成方法和作用,以期使得该类药物更加安全、有效。
关键词 仑伐替尼 衍生 体外活性
正文
肝癌是全球第三大癌症死亡的主要原因,其主要是由乙型肝炎病毒或丙型肝炎病毒的慢性感染、酗酒及代谢综合征(糖尿病和肥胖症)引起的[1-2]。中国是肝癌大国。据统计,全世界每年因肝癌造成70多万人死亡;仅在我国,每年的新发病例数及死亡病例数占全世界总例数的50%以上[3-4]。
根据恶性肿瘤细胞起源,原发性肝癌主要分为肝细胞癌(HCC)、肝内胆管细胞癌(ICC)和HCC-ICC混合型3种。其中,肝细胞癌(hepatocellular carcinoma,HCC)占所有原发性肝癌的85%-90%,是我国最常见的恶性肿瘤之一[3-4]。它是一种具有高度血管依耐性的实体肿瘤。HCC发生过程中,血管内皮生长因子受体(Vascular Endothelial Growth Factor Receptor,VEGFR)和血小板衍生生长因子受体(Platelet-derived Growth Factor Receptor,PDGFR)异常,可使RAS/MAPK通路下游上调,导致血管生成增加和细胞增殖,与HCC和转移相关。
乐伐替尼(Lenvatinib,E7080),一个具有多靶点的小分子酪氨酸酶受体抑制剂,主要靶点有VEGFR1-3,FGFR1-4,RET,c-KIT等。乐伐替尼于2015年2月获FDA批准用于晚期放射性-碘难治性分化型甲状腺癌的治疗。2018年,FDA证实批准乐伐替尼用于初治晚期不可手术的肝细胞肝癌的治疗[5-6]。相隔不到一个月,中国CFDA正式确认乐伐替尼在国内获批上市,用于晚期肝癌的一线治疗。虽然研究证实乐伐替尼药效要好于索拉菲尼,但乐伐替尼仍存在缺陷[7]。
乐伐替尼开发之初,其目的是用于甲状腺癌的治疗,并不是针对HCC开发。体外研究表明,其作用于VEGFR效果较好,但作用于PDGFR效果较弱。作用于VEGFR2-3比作用于FGFR1、PDGFR选择性高10倍。另外,由于乐伐替尼游离态(结构式如式①)在各种常用溶剂中均很难溶,合成其甲磺酸盐时,选用醋酸为溶剂,然后加入甲磺酸成盐(参考日本卫材专利WO2016/031841),该工艺不可避免的会导致基因毒性杂质的出现(例如式②等)。合成乐伐替尼时,只能尽可能的减少基因毒性杂质,而难以彻底除去基因毒性杂质。
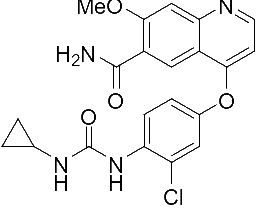
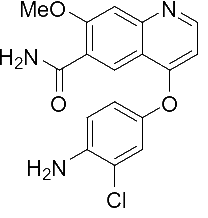
式① 式②
口服乐伐替尼不良反应较多,如高血压、腹泻、食欲降低、体重减轻和疲劳等。如此多的副反应,也可能与乐伐替尼各种基因毒性杂质太多有关[7]。
针对以上缺陷,本发明对乐伐替尼的结构进行了改良,以期望其针对肝细胞癌能发挥出更好的药效,同时减少药物中基因毒性杂质,降低药物副作用。因此,我们提出一种酪氨酸激酶抑制剂及其应用,以便于解决上述中提到的问题。
对此我们设计出仑伐替尼的衍生药物,为化合物A和化合物B。
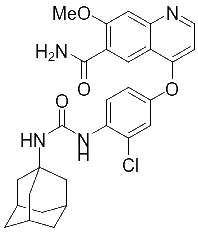
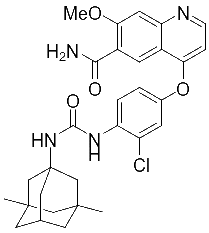
化合物A 化合物B
一、制备衍生药物
首先对化合物A和化合物B进行制备,两者的制备方法一致。以化合物A为例:
化合物A的制备方法,如图1中所示;
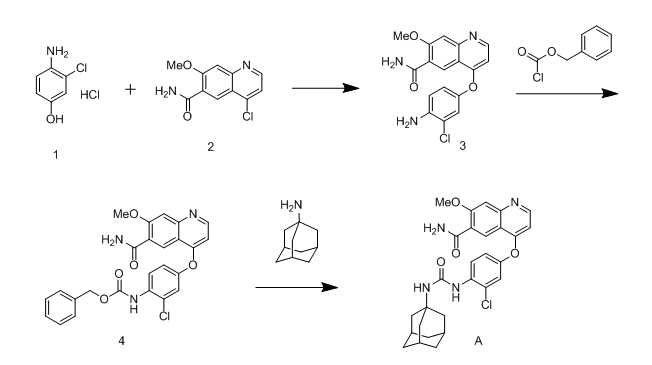
图 1
化合物3的合成
将900ml DMSO加入3L反应瓶,再加入64g KOH,抽真空,氮气置换一次,并保持通氮气下,向反应瓶中加入98.9g 化合物1。
用100ml DMSO冲洗粘壁物料。将反应液冷却20-25℃,向反应瓶中滴加水(滴加时间10-15分钟),(反应略有放热)控制温度小于40℃。
水滴加完后,保持搅拌,待体系由红棕色变成暗黑色(约0.5-1h),体系变色后再继续搅拌10-15分钟。
一次性将100g 化合物2加入到反应瓶中,升温至90℃,搅拌反应2小时,降温至55℃,依次向反应液中滴加500ml丙酮和1000ml水,滴毕后保温50至55℃搅拌30分钟。降温至30-35℃,再冰水浴降至5-10℃,保温搅拌30分钟。
抽滤,滤饼用100ml丙酮/200ml水的混合溶液洗涤,再用水洗涤滤饼至滤液基本无色;90℃以下,真空/鼓风干燥至水分小于0.2%,得类白色至红黄色固体(化合物3)132.0-138.0g,收率:91-95%;
液相检测,若单一杂质≥0.2%,则进行纯化,纯化步骤如下:将粗品132g加入到10ml/g THF中,加热回流2-3h,降温至10-20℃,过滤,滤饼用少量THF淋洗。60-80℃真空烘干,得成品118.8g,收率:85-90%。
化合物4的合成
称取130g化合物3加入到氮气保护下的3L三口瓶,加1300ml DMF,加65.8g吡啶,5.4g水,搅拌,降温至0-5℃,滴加142.0g氯甲酸苄酯到反应瓶中,滴毕,0-10℃反应。
滴加完后约30分钟,体系会析出固体,体系呈粘稠状。
反应约3小时后,向反应液中滴加1300ml水,控温小于等于20℃滴加,(刚加水时会放热)滴毕,搅拌0.5小时,控温10-20℃,抽滤,滤饼用520ml水洗涤。
再用520ml丙酮洗涤,60℃-70℃真空干燥至水分小于0.5% ,得白色或类白色固体(化合物4)162g,收率约90%。
化合物A的合成
称取125.7g 盐酸金刚烷胺,92.6g 碳酸钾加入到氮气保护下的3L三口瓶,加800ml DMF,加热至50℃搅拌1小时,加入53g吡啶,然后加入160g 化合物4,50℃搅拌反应约2小时后。向反应液中滴加1600ml水,控温小于等于20℃滴加,(刚加水时会放热)滴毕,搅拌0.5小时,控温10-20℃,抽滤,滤饼用1120ml水洗涤,再用560ml乙醇洗涤。60℃-70℃真空干燥至水分小于0.5% ,得白色或类白色固体(化合物二)157g。收率约90%。
精制
将所得粗品加入800ml乙醇回流1小时,冷却过滤即得纯品。
MS:m/z(ES):521.2,522.2[M+1]。
1H-NMR:
(400MHz ,d6-DMSO):8 .67(d ,2H) ,8 .28(d,1H) ,8.01(s ,1H) ,7 .86(s ,1H) ,7.75(s ,1H) ,7.51(s ,1H) ,7.47(d ,1H) ,7.18~7.21(dd ,1H) ,6.87(s ,1H) ,6.52(d ,1H) ,4.03(s ,3H),2.03(s ,3H),1.96(s ,6H),1 .64(s ,6H)。
化合物B的合成
制备流程参考化合物A的合成;将盐酸金刚烷胺替换成盐酸美金刚,可制得化合物B。
MS:m/z(ES):549.2,550.2[M+1]。
1H-NMR:
(400MHz ,d6-DMSO):8 .67(d ,2H) ,8 .27(d,1H) ,8.00(s ,1H) ,7 .86(s ,1H) ,7.75(s ,1H) ,7.51(s ,1H) ,7.47(d ,1H) ,7.18~7.21(dd ,1H) ,6.90(s ,1H) ,6.51(d ,1H) ,4.03(s ,3H),2.10(s ,1H),1.78(s ,2H),1.60(s ,4H),1.35(d ,2H),1.28(d ,2H),1.12(s ,2H),0.83(s ,6H)。
化合物A盐酸盐的制备
称取10g化合物A,加入300ml乙醇溶清,滴加10ml 15%的氯化氢乙醇溶液,滴毕,室温搅拌2小时,过滤,乙醇洗涤滤饼,真空干燥得约9g白色固体。
化合物B盐酸盐的制备
制备流程参考化合物A盐酸盐,可制得化合物B的盐酸盐。
二、纯度实验
1、实验条件
检测器:紫外吸收计(测量波长:252nm);
柱子:WONDASIL C18 SUPERB 5um 4.6MM*250MM;
柱温:恒温接近40℃;
流动相:如表1所示,用线性梯度洗提具有以下组成的溶液A和溶液B;
溶液A:水/乙腈/70.0~72.0% 分析纯高氯酸 (990:10:1,V/V/V);
溶液B:水/乙腈/70.0~72.0% 分析纯高氯酸 (100:900:1,V/V/V);
流速:1.0ml/min
时间(min) | 在流动相中溶液B的比例(vol%) |
0 | 5 |
15 | 40 |
25 | 60 |
35 | 100 |
40 | 100 |
40.01 | 5 |
45 | 停止 |
【表1】
实验原理
采用外标法检测文中实施例3或4制备的化合物A或B的盐酸盐和采用专利WO2016/031841方法制备的式①甲磺酸盐中式②的含量。
3、实验结果
名称 | 杂质式②的含量(按质量计) |
化合物A盐酸盐 | 未检出 |
化合物B盐酸盐 | 未检出 |
式①甲磺酸盐 | 78ppm |
【表2】
由表2可知,化合物A和化合物B盐酸盐,未检测出上文所述基因毒性杂质式②。而乐伐替尼甲磺酸盐现有工艺不可避免的会出现基因毒性杂质式②。
三、体外酶活性抑制测试
1、测试原理
通过Envision检测Assay plate中的化学发光,以化合物的IC50值为指标,来评价化合物对酪氨酸激酶的抑制作用。
2、测试方法
本体外酶活性抑制测试筛选了VEGFR2酶,VEGFR3酶,PDGFRα酶以及DNA-PK酶,四种酶对化合物A,B进行测试。
以PDGFRα酶为例,在激酶缓冲液中稀释酶、底物、ATP和抑制剂。添加1μl抑制剂(5%DMSO)、2μl酶(2ng)、2μl底物/ATP混合物(25uM ATP,0.2μg/μlPolyE4Y1)到384孔低容量微孔板中,在25℃孵育120分钟。加入5μlADP-Glo™试剂,在25℃孵育40分钟。加入10μl激酶检测试剂,在25℃孵育30分钟。记录发光值(积分时间0.5秒)。激酶缓冲液:40mM Tris,pH 7.5;20mM氯化镁;0.1mg/ml牛血清白蛋白(BSA);50μMDTT。
化合物检测8个浓度梯度,最高浓度为10µM,依次往下做5倍稀释。化合物浓度排布图(layout)如下【表3】。
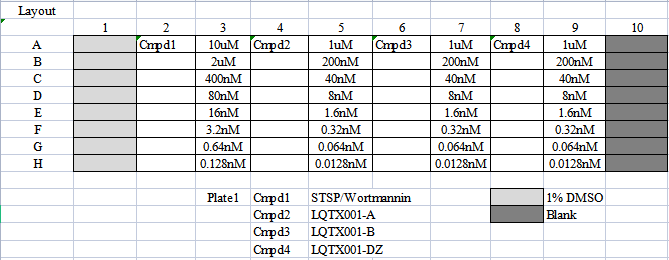
【表3】
LQTX001-A对应上文所述化合物A,LQTX001-B对应上文所述化合物B,LQTX001-DZ对应已上市乐伐替尼。STSP/Wortmannin为阳性药。VEGFR2酶,VEGFR3酶,PDGFRα酶三种酶对应STSP阳性药,DNA-PK酶对应Wortmannin阳性药。
测试数据:
测试四种酶的原始数据及%酶活率分别如下【表4,5,6,7】。
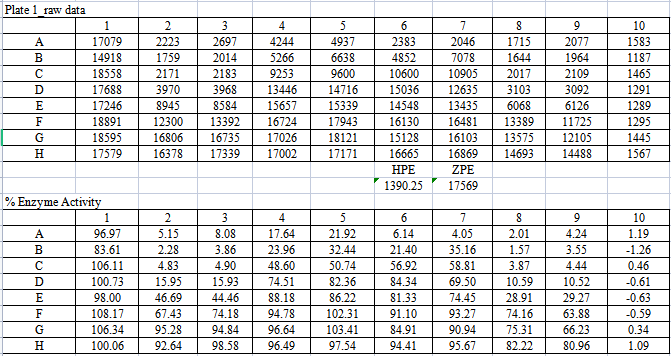
【表4:VEGFR2酶的作用数据及%酶活率】
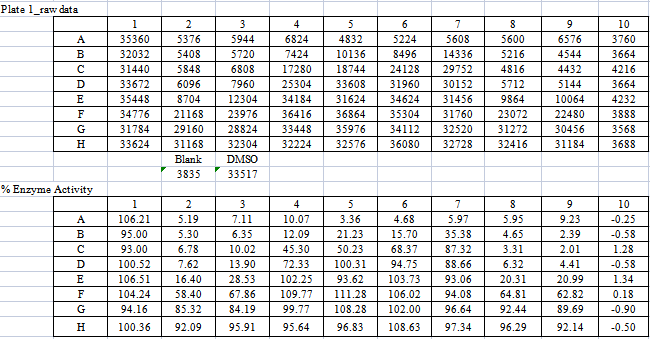
【表5:VEGFR3酶的作用数据及%酶活率】
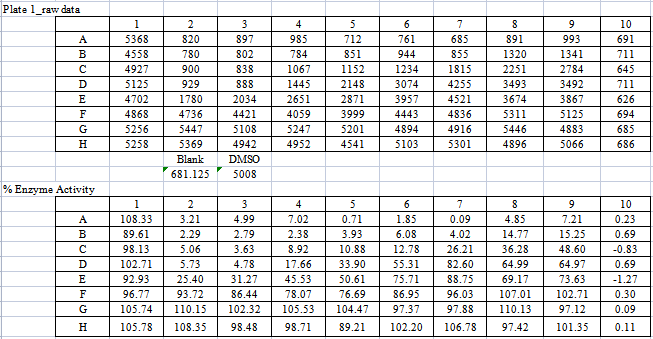
【表6:PDGFRα酶的作用数据及%酶活率】
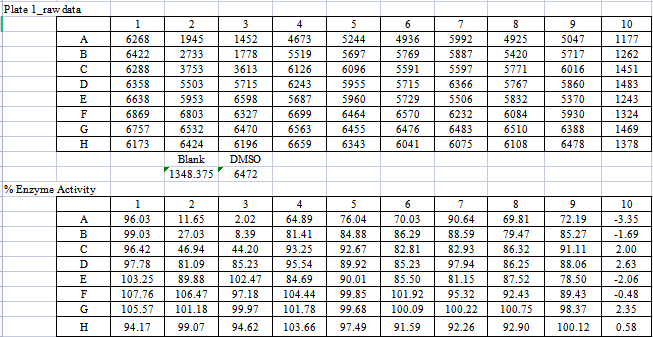
【表7:DNA-PK酶的作用数据及%酶活率】
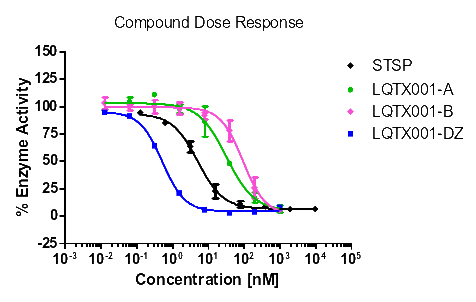
统计上述所有数据,更直观的酶活性和浓度梯度的线性图如下【图1,2,3,4】。
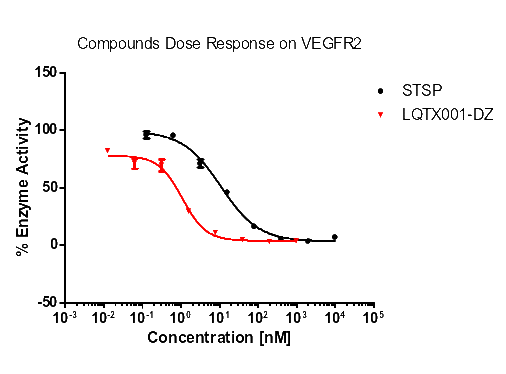
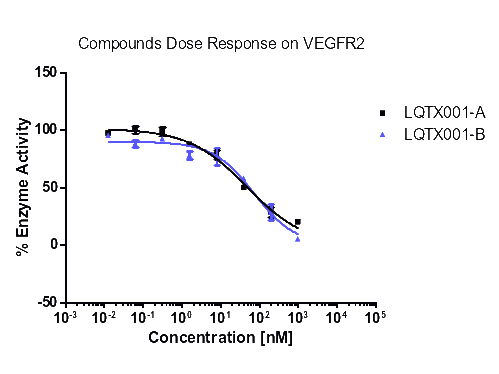
【图1:VEGFR2酶】
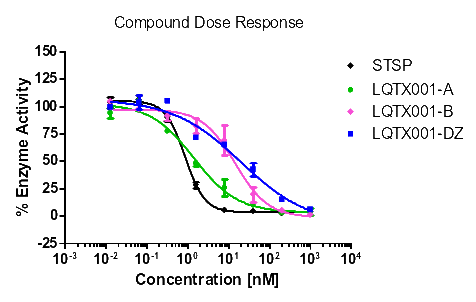
【图2:VEGFR3酶】 【图3:PDGFRα酶】
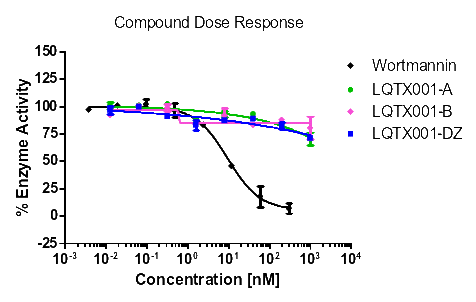
【图4:DNA-PK酶】
试验结论
名称 | VEGFR2 IC50(nM) | VEGFR3 IC50(nM) | PDGFRα IC50(nM) | DNA-PK IC50(nM) |
化合物A | 47.28 | 31.99 | 1.04 | >1000 |
化合物B | 72.59 | 92.68 | 14.39 | >1000 |
对照(式①) | 1.08 | 0.52 | 20.05 | >1000 |
【表8:化合物A,B对体外酶活性的不同抑制作用】
由表8可知,化合物A和B对试验中的酪氨酸激酶均具有抑制作用,尤其是对PDGFR具有更好的抑制作用,且对PDGFRα激酶的抑制作用优于对照式①(乐伐替尼),尤其是化合物A。
综上所述,我们发现该类仑伐替尼衍生药物具有潜在的研究价值,尤其是化合物A,同时由于研究时限的问题我们还未对其他靶点进行研究,如RET,c-KIT,MET,TAK-TAB等都可进行研究,该类衍生药物改变了靶点的特异性研究方向,从VEGFR转向了PDGFR。从结构上,我们可以尝试在金刚烷胺醇替代金刚烷胺,增加其亲水性。从体外活性测试上,我们也会进一步研究其他靶点的抑制活性以及其细胞毒性,以论证该类衍生物具有更好的潜在价值。
参考文献
[1] HUNYADY B, GERLEI Z, GERVAIN J, et al, Screening, diagnosis, treatment, and follow up of hepatitis C virus related liver disease. National consensus guideline in Hungary from 22 September 2017[J]. Orvosi Hetilap, 2018, 159(suppll 1):3-23.
[2] WANG L, LIU Y, ZHOU W, et al. Treatment related sever and fatal adverse events with molecular targeted agents in the treatment of advanced gastric cancer: a ment-
Analysis[J]. Onco Targets and therapy, 2017, 10:2281-2287.
[3]CHEN W, ZHENG R, BAADE PD, et al. Cancer statistics in China, 2015[J]. CA Cancer J Clin, 2016, 66(2): 115-132. DOI: 10.3322/caac.21338.
[4]GORDAN JD, KENNEDY EB, ABOU-ALFA GK, et al. Systemic therapy for advanced hepatocellular carcinoma: ASCO guideline[J]. J Clin Oncol, 2020, 38(36): 4317-4345. DOI: 10.1200/JCO.20.02672.
[5]Nakazawa Y, Kawano S, Matsui J, et al. Multitargeting strategy using Lenvatinib and golvatinib: Maximizing anti-angiogenesis activity in a preclinical cancer model[J]. Cancer science, 2015, 106(2):201-207.
[6] Wiegering A, Korb D, Thalheimer A, et al. E7080(Lenvatinib), a multi-targeted tyrosine inhibitor, demonstrates antitumor activites against colorectal cancer xenografts[J]. Neoplasia(New York, NY), 2014, 16(11):972-981.
[7] 玉井俊行.肝细胞癌的治疗:日本,CN 110831597 A[P].2020-02-21:71-73.
8

客服QQ:30444492琼网文【2021】1550-113号
增值电信业务经营许可证:琼B2-20210322
出版物经营许可证:新出发龙华出字第(2021)009号
广播电视节目制作经营许可证:(琼)字第00779号
版权所有 ©2002-2024 期刊网(www.qikanchina.com) 琼ICP备2021005105号



